The direct conversion of carboxylic acids or acid chlorides or esters to ketones or aldehydes using organometallic reagents usually does not result in high yield because the intermediate ketones are highly reactive towards the organometallic reagents.
However, after derivatization of these carboxylic acid derivatives to the corresponding Weinreb amide, the reaction with organometallics does give the desired ketones in high yield because the initial adduct is stabilized and does not undergo further reaction. A nucleophilic addition to the Weinreb amides results in a stable five-membered cyclic tetrahedral intermediate which protects the over-oxidation.
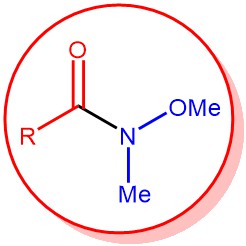
N-Methoxy-N-methyl amides are called Weinreb amides. The Weinreb amide can be easily prepared from activated carboxylic acid derivatives (e.g., acid chloride or anhydride) and N,O-dimethylhydroxylamine hydrochloride in the presence of a base. On the other hand, esters or lactones can be converted to the corresponding Weinreb amide using trimethylaluminum (Me3Al) or dimethyl laluminum chloride (Me2AlCl).

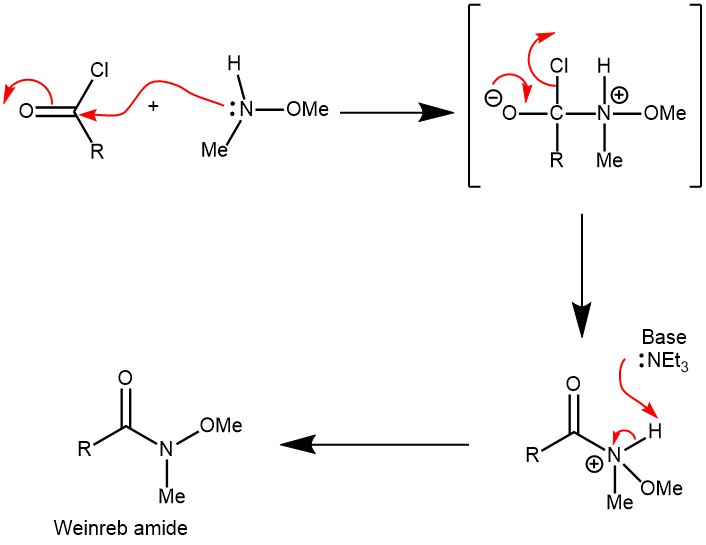
MECHANISM:FORMATION OF WEINREB’S AMIDE
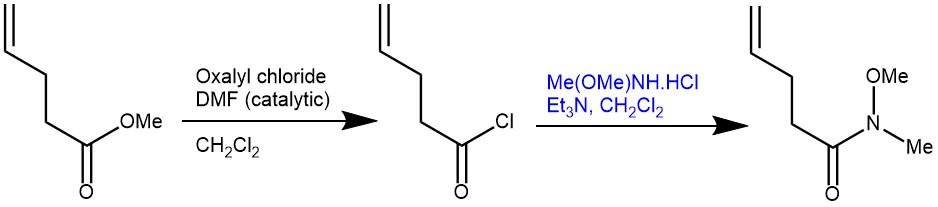
EXAMPLE 1: N-Methoxy-N-methylpent-4-enamide. A 1 L round-bottomed flask equipped with a stir bar is loaded with 4-pentenoic acid (25.5 mL, 249.9 mmol, 1.0 equiv) and CH2Cl2 (500 mL). The resulting solution is magnetically stirred (500 rpm) and oxalyl chloride (22.2 mL, 262.3 mmol, 1.05 equiv) is added dropwise at 24 °C via a 50 mL dropping funnel. The addition is complete within 5 min, at which time the dropping funnel is removed and DMF (0.1 mL) is added to the reaction mixture in one portion with a syringe. The solution begins to vigorously bubble. The reaction flask is quickly connected in series, first to an empty wash bottle and then to a wash bottle filled with a 5 M NaOH solution (200 mL). The colorless reaction mixture is stirred at 24 °C for 2 h, during which bubbling ceases. The reaction mixture is concentrated on a rotary evaporator (40 °C, 375 then 110 mmHg) leaving the stir bar inside the flask (Note 7). The remaining orange oily residue is dissolved in CH2Cl2 (250 mL) and again concentrated on a rotary evaporator (40 °C, 375 then 110 mmHg). The residue is then dissolved in CH2Cl2 (400 mL), cooled to 0 °C in an ice bath, and N,O-dimethylhydroxylamine hydrochloride (24.35 g, 249.7 mmol, 1.0 equiv) is added in one portion to make a suspension. The flask is equipped with a 100 mL dropping funnel filled with triethylamine (72.7 mL, 524.4 mmol, 2.10 equiv). Triethylamine is added dropwise within 5 to 10 min to the magnetically stirred (500 rpm) reaction mixture at 0 °C. After the addition is complete, the reaction mixture is stirred at 0 °C for 2 h and a white suspension forms. The suspension is poured into a 1 L separatory funnel containing a saturated solution of NaHCO3 (400 mL) at 24 °C. The reaction flask is rinsed with CH2Cl2 (2 x 25 mL) and the resulting solutions are added to the separatory funnel. After shaking, the phases are separated and the yellow-orange organic layer is further washed with water (2 x 300 mL) and a saturated aqueous NaCl solution (300 mL), dried over MgSO4 (50 g), filtered through fluted filter paper in a glass funnel, and concentrated on a rotary evaporator (40 °C, 500 then 15 mmHg). The residual orange-colored oil (contaminated with solid Et3N•HCl) is transferred into a 100 mL round-bottomed flask (NS24/40) equipped with a 2.5 cm long oval Teflon coated stir bar. This is connected to a short distillation bridge (Liebig condenser) equipped with a thermometer and a distillation cow bearing four 100 mL flasks. The distillation flask is submerged into an oil bath on a magnetic hot plate and is covered with aluminum foil from above the oil level up to the thermometer. The apparatus is connected via high vacuum-grade silicone tubing to a Schlenk-line and evacuated. By using a needle valve, a pressure of 7–10 mmHg is maintained. After waiting 2–3 min until residual solvent removed (indicated by ceased bubble formation) the oil bath temperature is gradually increased from 24 °C to 95 °C. The pure product distills at a is temperature of 85 °C (oil bath temperature: 95 °C) and a pressure of 7–10 mmHg (Note 15). Weinreb amide 1 is obtained as a colorless oil in a yield of 69% (24.69 g, 172.4 mmol) [REF: Org. Synth. 2021, 98, 171-193; DOI: 10.15227/orgsyn.098.0171]
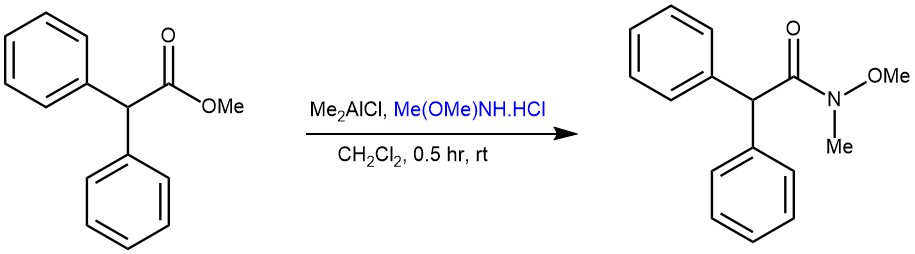
EXAMPLE 2: A solution of Me2AICl (1.01 M hexane solution, 2-5 equiv) was added over a 5-min period to a stirred suspension of MeONHMe.HCl (the same equiv as that of Me2A1CI) in CH2C12 (20 ml) under nitrogen at 0 °C, and the mixture was stirred for 1 h allowing the temperature to rise to room temperature. Then, a solution of lactone or ester (! mmol) in CH2C12 (10 ml) was added dropwise. After the reaction was completed, a solution of a phosphate buffer (pH 8.0) (3 ml per 1 mmol of Me2AIC) was added and the stirring was continued for 10 min. The mixture was diluted with CHCI3, filtrated through a Celite pad, and washed thoroughly with CHC13. The aqueous layer was extracted with CHCI3. The combined organic layers were washed with brine, dried over MgSO4, and concentrated. The residue was purified by silica gel flash column chromatography to give the amide.[REF: Tetrahedron Letters, Vol. 38, No. 15, pp. 2685-2688, 1997 ]
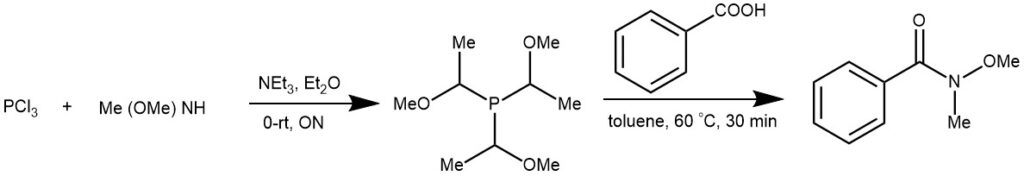
EXAMPLE 3: A solution of PCl3 (20mmol, 2.74 g) in dry Et2O (25 mL) was added at 0°C dropwise into a stirred mixture of Me(MeO)NH (62mmol, 3.78g) and NEt3 (70 mmol, 20ml) in dry diethyl ether (50 mL) under nitrogen. The reaction mixture was then allowed to warm to room temperature slowly and stirred overnight. The mixture was refluxed for another 4h. After cooling to room temperature, the reaction mixture was filtered and the filtrate was concentrated in vacuum to afford product (2.81g, 67%) as colorless oil.
Compound P[NCH3(OCH3)]3(1mmol)and benzoic acid (0.244g, 2 mmol) were dissolved in toluene (10 mL) in a 50 mL three-necked fask under nitrogen. The mixture was stirred at 60 °C for 30 min, and the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature. The reaction was then quenched with saturated NaHCO3 solution (15 mL) and extracted with ethyl acetate (3×10 mL). The combined organic layers were dried over MgSO4. The solvent was removed under vacuum. The product was purified by column chromatography on silica gel (petroleum : ethyl acetate = 2:1 ) to give 5a in a 95% yield. [REF: Org. Lett., Vol. 11, No. 19, 2009]
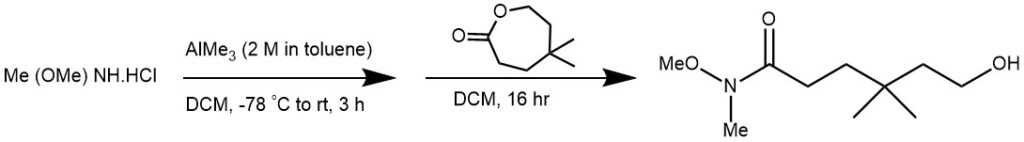
EXAMPLE 4: Trimethylaluminum (2 M in toluene, 3.75 mL, 7.5 mmol) was added dropwise over 20 minutes to a solution of N,O-dimethylhydroxyl amine hydrochloride (0.79 g, 8.1 mmol) in DCM (5 mL) at -78 C. The solution was allowed to warm to room temperature and was stirred for 3 hours followed by cooling to -10 C and slow, dropwise addition of 5,5-dimethyloxepan-2-one (0.5 g, 3.5 mmol) as a solution in DCM (1 ML). The reaction mixture was stirred at room temperature for 16 hour before careful, dropwise addition of Rochelle’s salt (1.19 g, 4.2 mmol) in water (10 mL). The resulting mixture was stirred for a further 16 hour before filtration and repeated washing with DCM (100 mL). The filtrate was then dried, filtered, and the solvent removed in vacuo. The crude product was further purified by flash column chromatography (25% ethyl acetate in petroleum ether) to get the product (0.68 g, 96%) as pale yellow oil. [REF: Org. Lett., Vol. 7, No. 7, 2005]
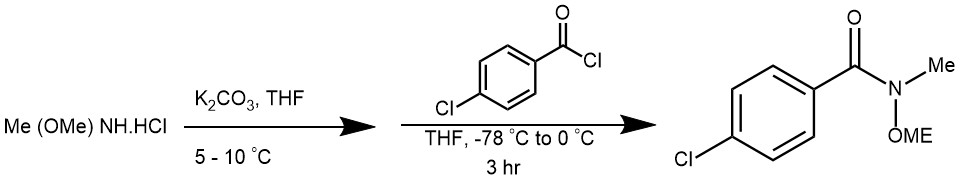
EXAMPLE 5: A solution of N,O-dimethylhydroxylamine was prepared in situ by stirring a suspension of N,O-dimethylhydroxylamine hydrochloride (8.36 g, 85.7 mmol, 1.5 equiv.) in THF (60 ml) for 30 min. Water (2.0 ml) was added to this suspension and this was stirred for a further 20 min. The temperature of the mixture was decreased to 5-10°C and potassium carbonate (18.0 g) was added. This was stirred for 30 min, maintaining the temperature between 5 and 10 °C. The mixture was then filtered and the solution of N,O-dimethylhydroxylamine thus obtained was added to a solution of p-chlorobenzoyl chloride (10.0 g, 57.1 mmol, 1.0 equiv.) in THF (60 ml) at -78°C. This was stirred for 3 h at -78°C and the temperature was allowed to warm to 0°C. Sodium bicarbonate was added and the phases were separated. The aqueous phase was extracted 3 times with diethyl ether. The combined organic fraction was washed with sodium bicarbonate, water, dried over magnesium sulfate, filtered and the solvent was removed in vacuo. The crude product was purified by column chromatography (hexanes – diethyl ether, 80:20 then 60:40) to afford the title compound 11 (11.39 g, 95% yield) as a pale yellow liquid. [REF: Org. Lett., Vol. 7, No. 7, 2005]
REFERENCES:
- Strategic applications of named reactions in organic synthesis by Laszlo Kurti and Barbara Czako
- KHALID et al., Orient. J. Chem., Vol. 35(6), 1611-1626 (2019)
- Weinreb ketone synthesis. (2023, July 22). In Wikipedia. https://en.wikipedia.org/wiki/Weinreb_ketone_synthesis
- https://mychemblog.com/part-ii-weinreb-ketone-synthesis-carbon-carbon-bond-formation-using-weinreb-amide-and-organometallic-reagents/

