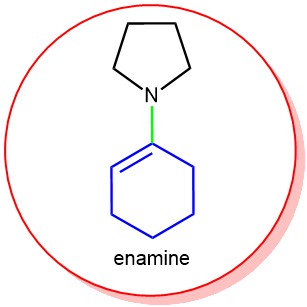
An indirect method for the selective alkylation and acylation of an aldehyde or ketone is via an intermediate called ‘Enamine’. An enamine is an unsaturated compound derived from the condensation of an aldehyde or ketone with a secondary amine. Enamine can be compared with ‘Enol’. Enamines are considered to be nitrogen analogs of enols.

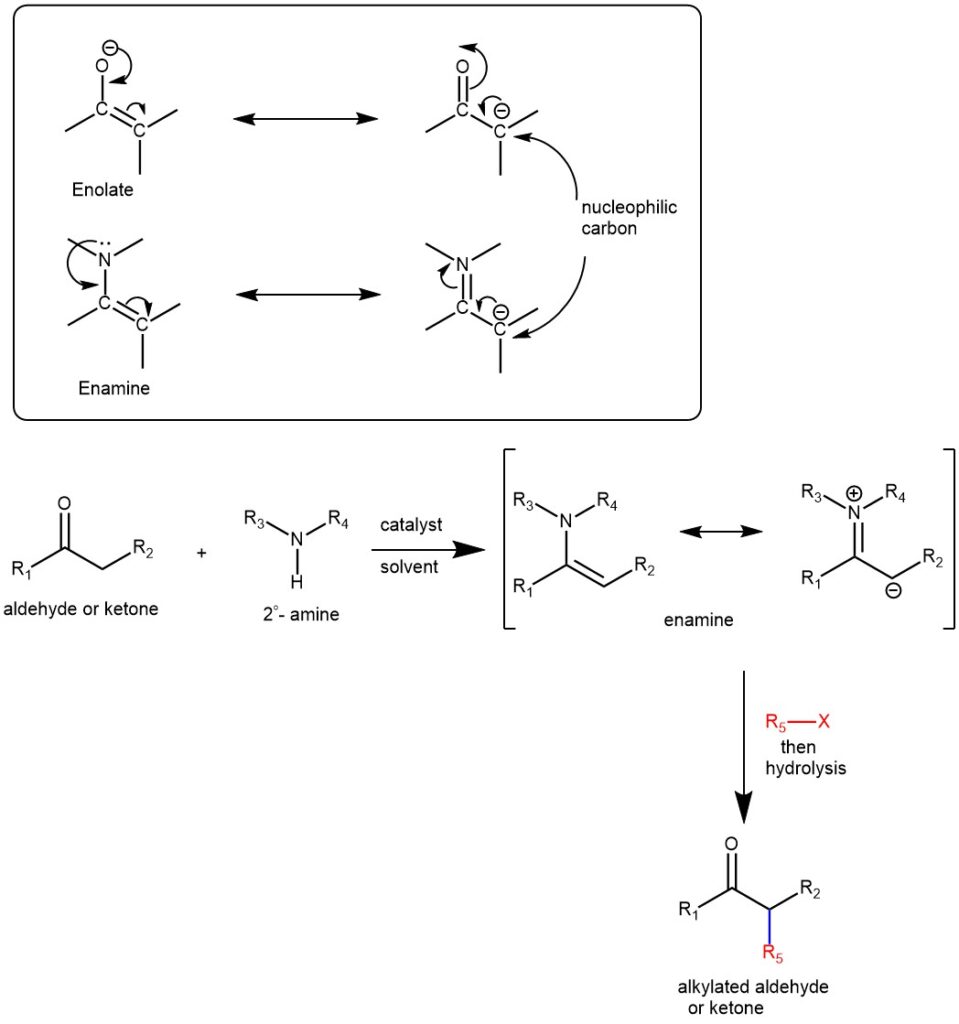
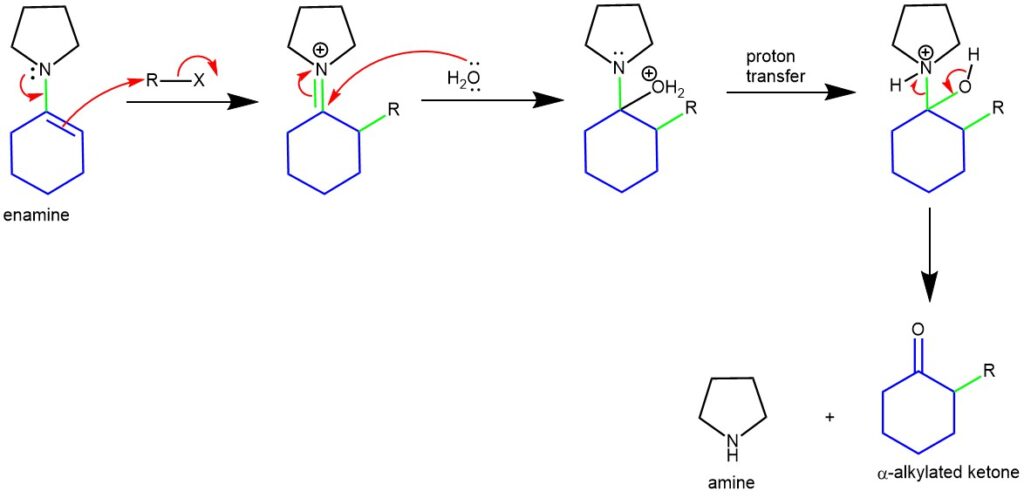
Enamines act as nucleophiles in a fashion similar to enolates. Because of this enamines can be used as synthetic equivalents of enolates. In 1954, G. Stork and co-workers discovered that the reaction of enamines with alkyl or acyl halides followed by acidic hydrolysis constitutes a novel way for the α-alkylation and α-acylation of carbonyl compounds (aldehyde or ketone). The synthesis of α-alkyl or α-acyl carbonyl compounds via the alkylation or acylation of the corresponding enamines is known as the Stork enamine synthesis. The electronic distribution in enamines shows that the beta-carbon atom bears an appreciable negative charge and serves as a nucleophile.
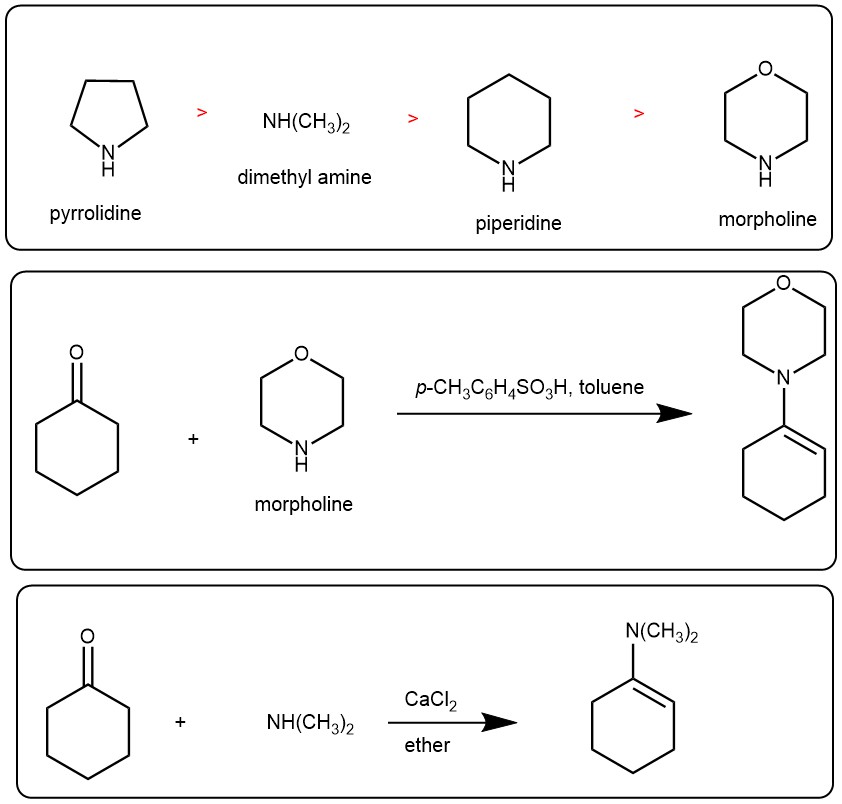
The enamines are prepared by reacting the aldehyde or ketone with one equivalent of secondary amine. Typically, the following 2°amines are used for the enamine reaction and the hydrolysis of the corresponding enamines follow the following order.

ALKYLATION: When enamines are treated with alkylating agents, the reaction leads to C-alkylation at the α-carbon ( with the possibility of N-alkylation). Activated alkyl halides are the best reaction partners (e.g., allyl, benzyl, propargylic, activated aryl halides, Michael acceptors, or epoxides). Tertiary alkyl halides do not alkylate the enamines (rather undergo elimination). Subsequent hydrolysis of the C-alkylated iminium salt yields an alkylated ketone or aldehyde.

MECHANISM OF ENAMINE FORMATION:
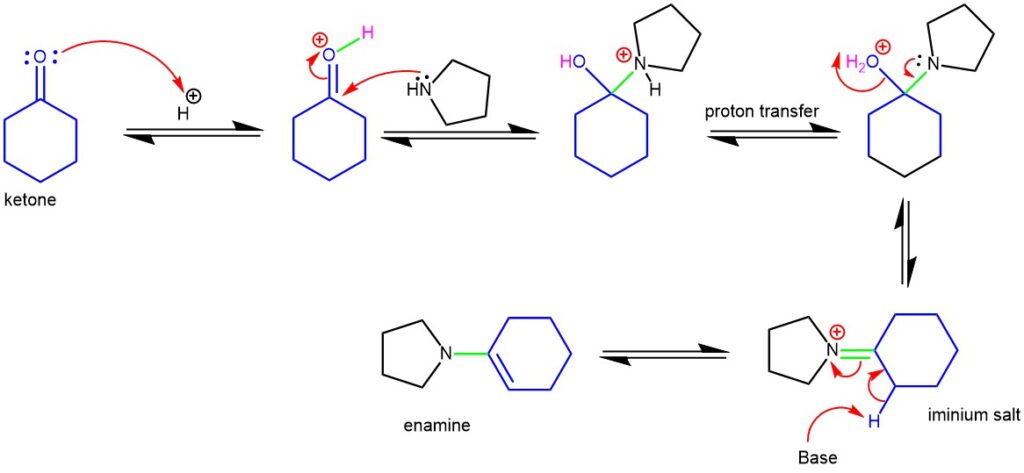
MECHANISM OF ALKYLATION OF ENAMINE AND HYDROLYSIS:
EXAMPLE 1: A solution of ketone 13 (1.0g, 5.60mmol, 1eq) and dry pyrrolidine (33.3mL, 71eq) in benzene (130mL) was heated under reflux at 100°C for 6h in an apparatus equipped with a water separator under argon. The solvent and pyrrolidine were then evaporated in vacuo. The oily residue (1.27g) and ethyl 2-bromoproprionate (1.2mL) were dissolved in dry dioxane (9.0mL) and heated under reflux at 120°C overnight under argon. Water (4.5mL) was added, and the mixture heated under reflux for a further 1h. After cooling to RT, the aqueous layer was extracted with Et2O (3 × 100mL), and the organic layers were combined, washed with water (250mL), brine (250mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification by flash chromatography (P.E.40-60: Et2O 4:1) gave the title compound 14 as a yellow oil consisting of a mixture of diastereomers (0.50g, 52% conversion yield) and recovered starting material 13 (0.44g). [REF: Org. Lett., Vol. 5, No. 17, 2003]

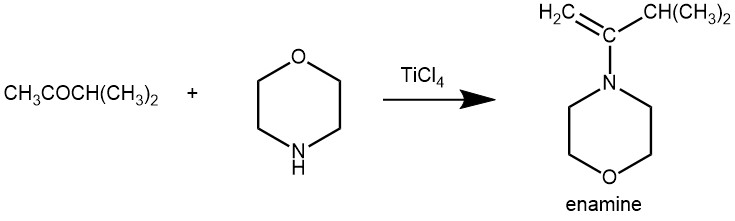
EXAMPLE 2: To a 2-1., four-necked flask fitted with a stirrer, condenser, N2 gas line, thermometer, and dropping funnel is added 500 ml of solvent (benzene, n-pentane, or ethyl ether). To this is added 0.1 mole of ketone and a solution of amine (excess) in 100 ml of solvent. To the resulting solution is added, over a 20-60-min period, 0.055 mole TiCl4 in an additional 100 ml of solvent (benzene or n-pentane). The temperature is kept between 0° and 10° during this addition. When the TiCl4 addition is complete, the mixture is allowed to stir at room temperature for several hours. The more hindered ketones required longer reaction times and the progress of the reaction is followed by nmr analysis of aliquots. The reaction mixture is then filtered and the solvent is removed. The residual oil is distilled at reduced pressure. All operations described above are carried out under an atmosphere of dry nitrogen. [REF: The Journal of Organic Chemistry 32.1 (1967): 213-214.]
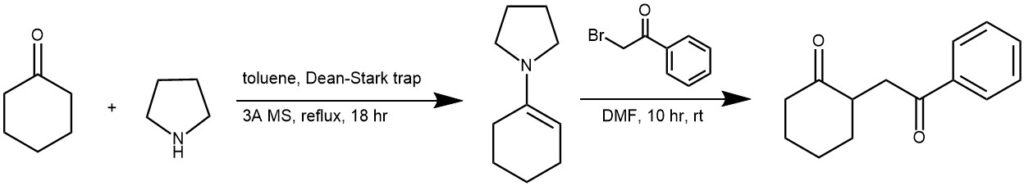
EXAMPLE 3: A 50 mL round-bottomed flask containing cyclohexanone (3.82 g, 39 mmol) in anhydrous toluene (20 mL) was fitted with a Dean-Stark trap containing 3A molecular sieves, reflux condenser, and a heating mantle. Pyrrolidine (6.00 mL) was added, and the solution was heated to reflux for 18 hours. The solvent was evaporated and the crude product 1-cyclohex-1-en-1-ylpyrrolidine (enamine) was used directly for the next reaction (6.0 g, 102% yield, 95% purity by HPLC)
To a 250 mL round-bottomed flask containing 2.4 mL of enamine was added 100 mL anhydrous DMF, under nitrogen. The flask was fitted with an addition funnel containing 2-bromo-1-phenylethanone (4.12 g) dissolved in 35 mL anhydrous DMF, which was dropped into the enamine solution over 60 min. This solution was stirred at ambient temperatures for 10 hours, then 90 mL of water was added to the solution and it was stirred for another 11 hours, under nitrogen. The solution was then extracted twice with ethyl acetate and the organic layers combined and further washed with water (3 times), dried over anhydrous sodium sulfate, filtered and evaporated to give a yellow oil. The oil was purified by flash chromatography using a gradient cyclohexane: ethyl acetate (9:1) to get the product (4.0 g, 90% yield, 98% purity by HPLC) [REF: World Intellectual Property Organization WO 2008/116926 A1]
REFERENCES:
- Strategic applications of named reactions in organic synthesis by Laszlo Kurti and Barbara Czako
- Modern organic synthesis by Herbert O. House
- Enamine. (2023, May 13). In Wikipedia. https://en.wikipedia.org/wiki/Enamine
- https://chem.libretexts.org/

