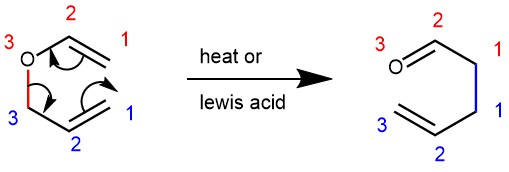
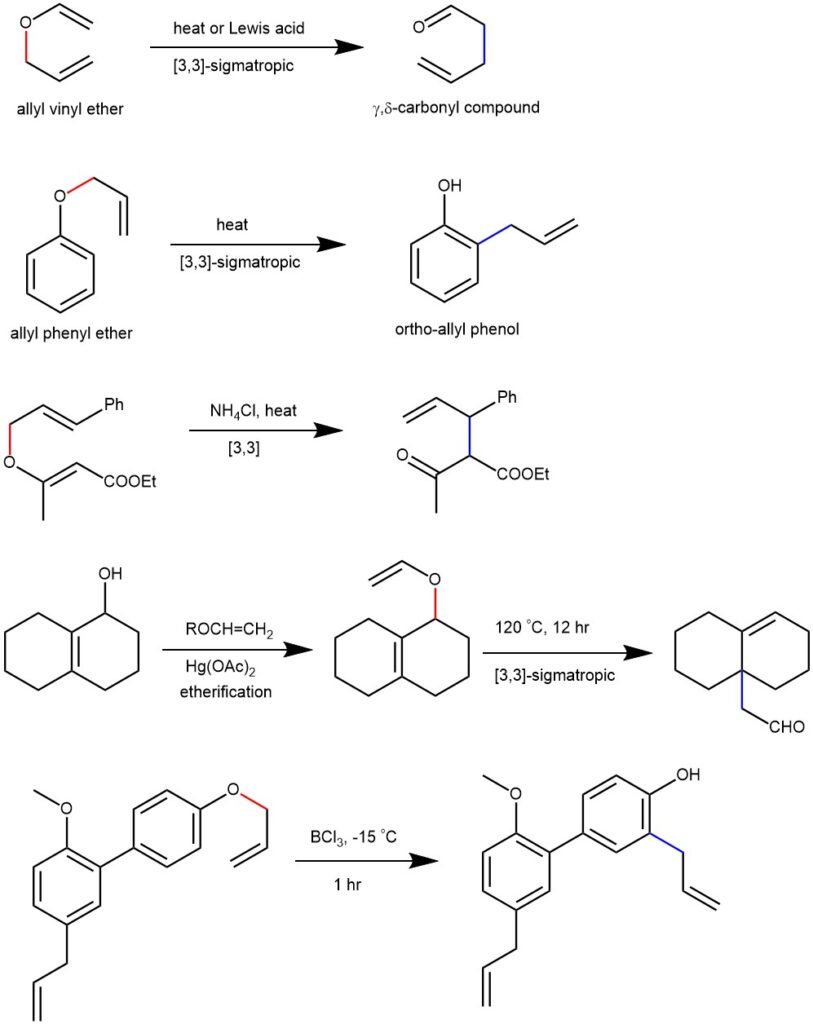
Thermal [3,3]-sigmatropic rearrangement of allyl vinyl ether (or allyl phenyl ether) to the corresponding unsaturated carbonyl compounds are called the Claisen rearrangement. It was first reported by Ludwig Claisen in 1972.
There are different variations of Claisen rearrangement like Bellus-Claisen rearrangement, Eschenmoser-Claisen rearrangement, Ireland-Claisen rearrangement, Johnson-Claisen rearrangement, Kazmaier-Claisen rearrangement, Aza-Claisen rearrangement, etc.
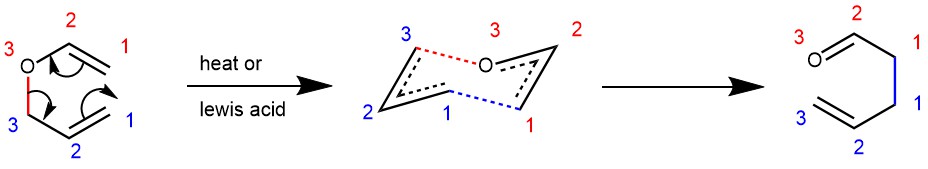
MECHANISM: Mechanistically the reaction can be described as a suprafacial, concerted, [3,3]–sigmatropic rearrangement. The reaction is unimolecular and proceeds preferably via a chair-like transition state.
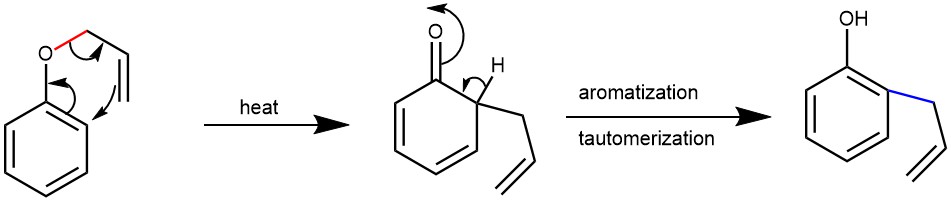
The aromatic Claisen rearrangement (allyl phenyl ether) is followed by rearomatization.
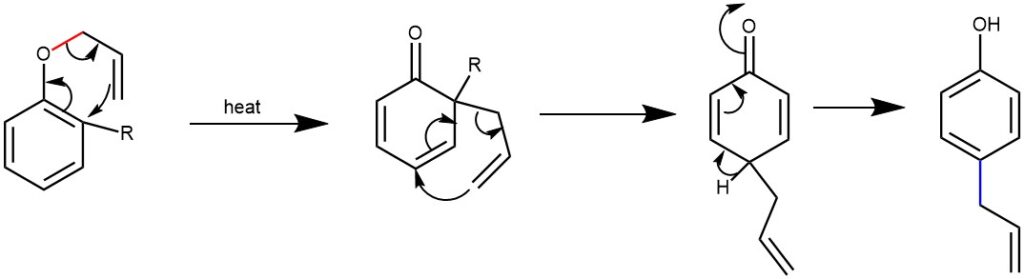
When the ortho position is substituted, rearomatization cannot take place. Therefore, the allyl group undergoes Cope rearrangement to the para-position before aromatization is possible.
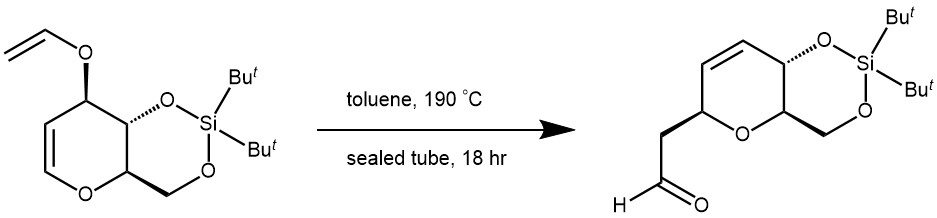
EXAMPLE 1: A solution of starting material (5.0 g, 16.02 mmol) in toluene (40 mL) was heated at 190 °C in a sealed tube for 18 h, solvent was removed under reduced pressure. The crude was purified via silica gel chromatography (10% EtOAc in hexane) to afford aldehyde (4.0 g, 80%) as a white solid. [REF: Org. Lett. 2018, 20, 96−99]
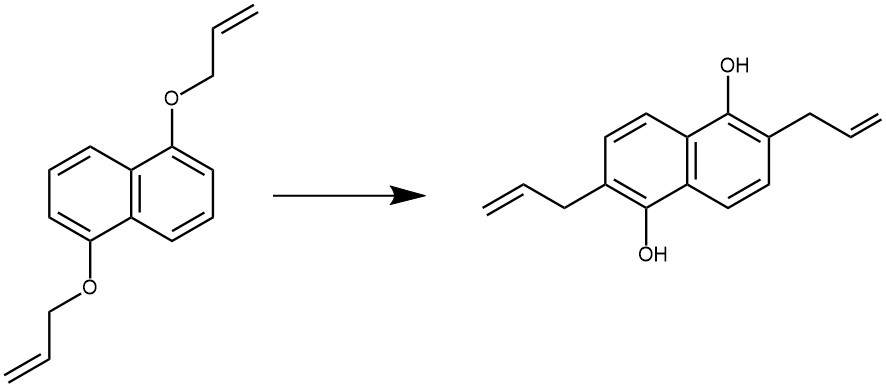
EXAMPLE 2: A 200 mL round-bottom flask was charged with 1,5-bis(allyloxy)-naphthalene (5.4 g, 22.5 mmol) and then was sealed with a septum stopper. The flask was fitted with a gas inlet and outlet and then flushed with nitrogen for ten minutes. The vessel was heated with constant agitation under a steady flow of nitrogen in a pre-heated 190 °C, mineral oil bath for ten minutes. The vessel was then allowed to cool to room temperature under a positive pressure of nitrogen. The rearranged Claisen product was isolated in 100% yield (5.4 g, 22.5 mmol).[REF: Milliken and company-US2006/223993, 2006, AI)
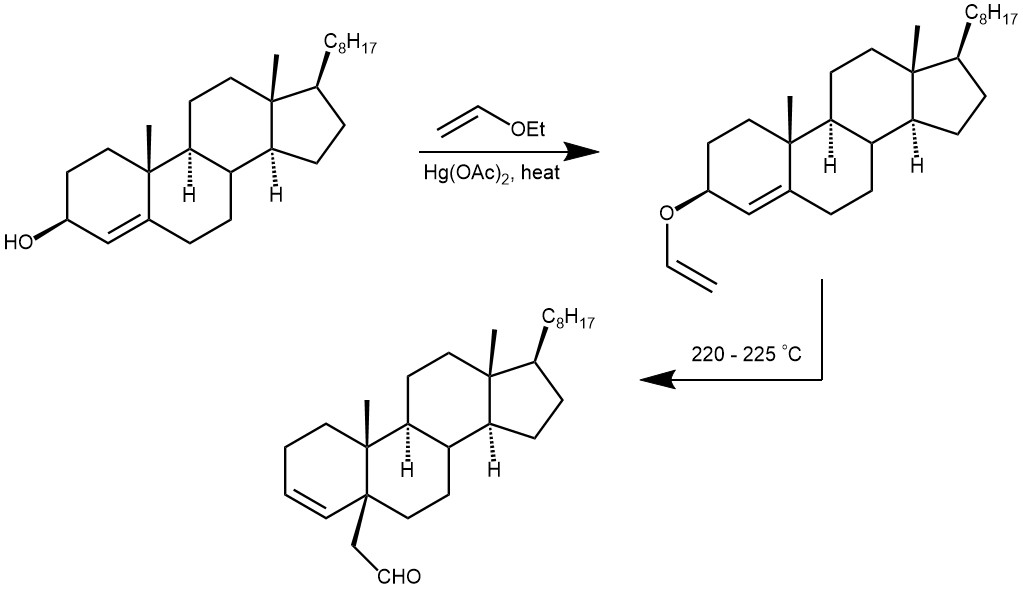
EXAMPLE 3: A 50-mL round-bottomed flask equipped with a magnetic stirring bar and a 20-Ml calibration mark is charged with 970 mg. (2.51 mmoles) of cholest-4-en-3β-ol. Ethyl vinyl ether is distilled into the flask to the 20-mL mark. The mixture is stirred to effect solution before 820 mg. (2.55 mmoles) of mercury (II) acetate is added to the reaction mixture. The flask is fitted with a reflux condenser connected to a gas-inlet tube and flushed with argon. The reaction mixture is then stirred and heated at reflux under a positive argon pressure for 17 hours. After the solution has cooled to room temperature, 0.062 ml. (1.1 mmoles) of glacial acetic acid is added, and stirring is continued for 3 hours. The reaction mixture is poured into a pre-shaken mixture of 150 mL of petroleum ether and 50 mL of 5% aqueous potassium hydroxide. The aqueous phase is extracted with 50 ml. of petroleum ether, and the combined extracts are washed with three 50-mL portions of a 20% aqueous sodium chloride, dried over anhydrous sodium carbonate, filtered, and evaporated at reduced pressure giving 1.11 g of an oil which, upon filtration through 5 g of silica gel with 200 ml. of petroleum ether, affords 0.81 g. of the cholestenyl vinyl ether as a clear, colorless oil. If desired, crystallization of this oil from 10 ml. of acetone will give 0.74 g. (71%) of the vinyl ether as colorless prisms, m.p. 55–56.5°.
The crude vinyl ether (0.81 g.) is transferred with petroleum ether into a 50-mL round-bottomed flask fitted with a long gas-inlet tube. After the petroleum ether is removed at reduced pressure the flask is filled with argon and heated under a positive argon pressure at 220–225 °C for 5 hours; little or no bubbling should occur. After cooling, the oil is chromatographed on 75 g. of silica gel using 10% diethyl ether in petroleum ether as the elution solvent. The first 175 ml. of eluant contains side products and is discarded; elution with another 175 ml. of the solvent gives 0.45–0.55 g. (50–53% overall yield from cholest-4-en-3β-ol) of 5β-cholest-3-ene-5-acetaldehyde as white prisms, m.p. 66.5–68 °C.[REF: Organic Syntheses, Coll. Vol. 6, p.298 (1988); Vol. 54, p.71 (1974)]
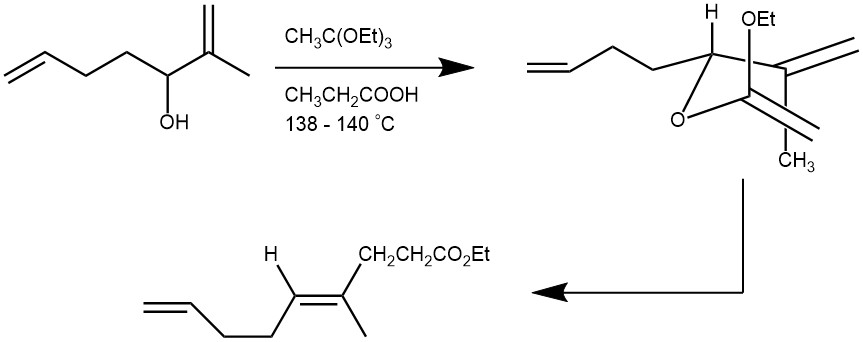
EXAMPLE 4: A 500-mL one-necked, round-bottomed flask containing a magnetic stirring bar is fitted with a Claisen adapter, two thermometers, and a receiving flask. To the flask is added 186 g. (1.15 moles) of ethyl orthoacetate 25.2 g (0.200 mole) of 2-methyl-1,6-heptadien-3-ol, and 0.70 g. (0.0094 mole) of propionic acid. The mixture is heated with stirring to keep the temperature above the liquid at 138–142°. Heating is continued until ethanol no longer distils from the reaction flask (approximately one hour is required). The reaction mixture is allowed to cool to room temperature, and the excess ortho ester and propionic acid are removed by distillation under reduced pressure (approximately 50–60° at 20 mm.). The colorless to yellow residual liquid is then distilled under reduced pressure (0.25 mm), giving 32.6–34.6 g. (83–88%) of ethyl 4-methyl-(E)-4,8-nonadienoate as a colorless liquid, b.p. 54–55° (0.25mm.).[REF: Organic Syntheses, Coll. Vol. 6, p.606 (1988); Vol. 53, p.116 (1973).]
REFERENCES:
- Strategic applications of named reactions in organic synthesis by Laszlo Kurti and Barbara Czako
- Tetrahedron Letters, 2009, 50, 1151-1152