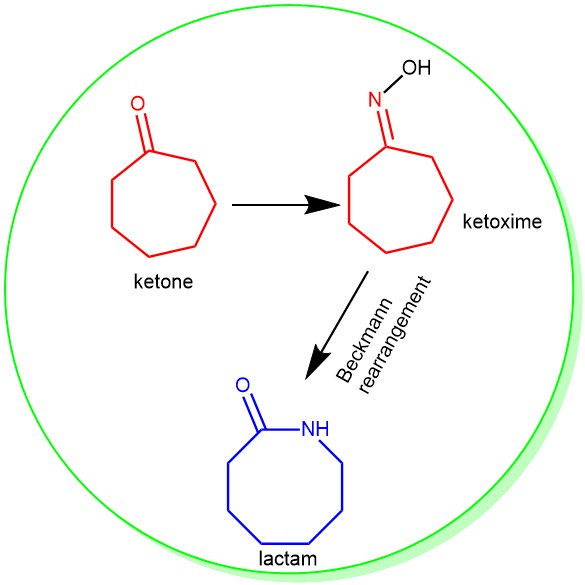
The rearrangement of ketoximes to the corresponding amide is known as the Beckmann rearrangement. It accomplishes in one stroke both the cleavage of a carbon-carbon bond and the formation of a carbon-nitrogen bond. This is achieved through the conversion of the oxime’s -OH group to a good leaving group (e.g., H2O, OTs, OMs, Cl, etc.) followed by heat. The reaction is usually carried out under temperatures greater than 130 °C and in the presence of a large excess of strong Bronsted acid such as sulfuric acid and acetic acid. However, converting the oxime’s -OH group into a much better-leaving group allows the reaction to be achieved at much milder conditions. The stereochemical outcome of this rearrangement is predictable: the R (alkyl) group anti to the leaving group on the nitrogen will migrate. The hydrogen atom never migrates, so this method cannot be used for the synthesis of N-unsubstituted amides.
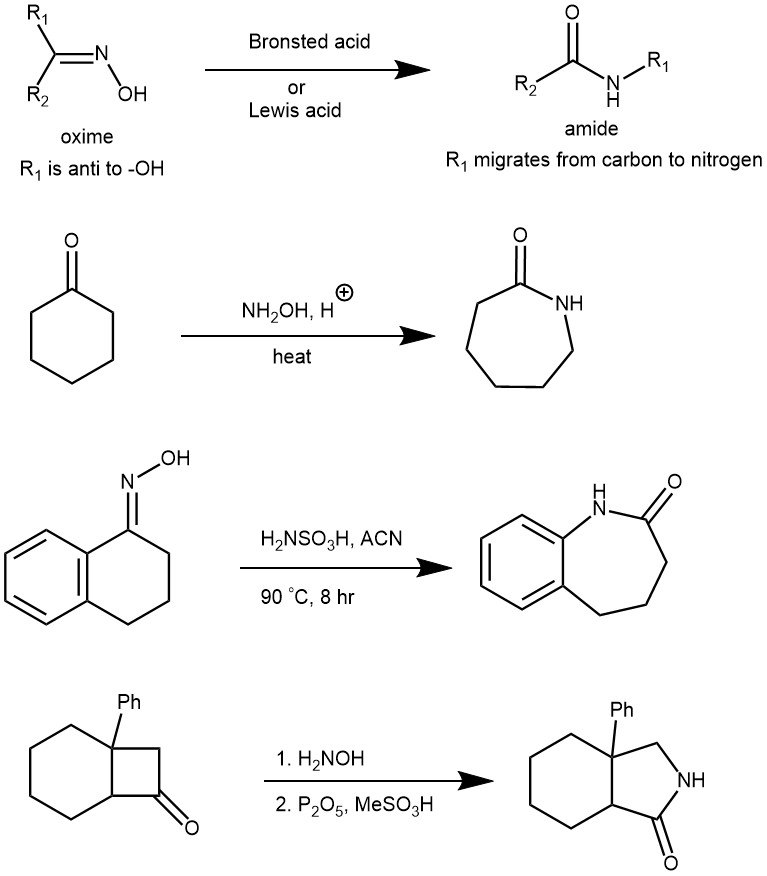
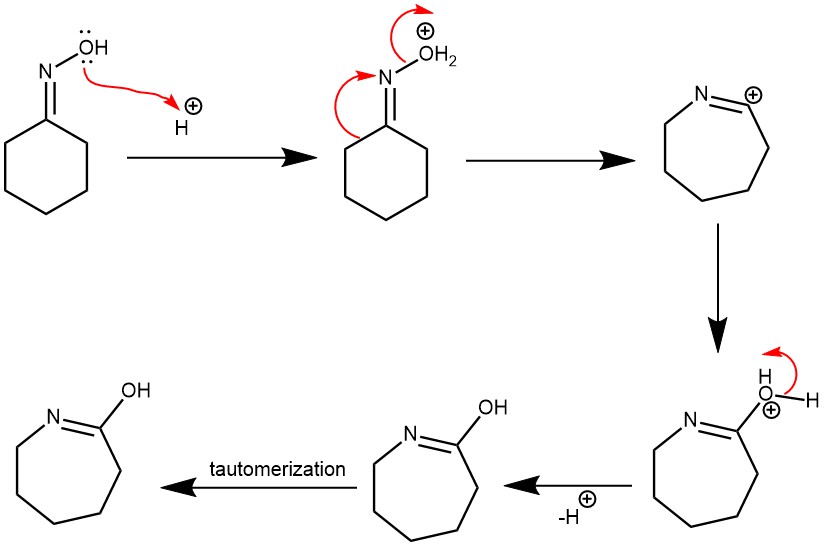
MECHANISM: When Bronsted acid (e.g., H2SO4) is used, the oxime oxygen is protonated followed by the loss of H2O (a better leaving group than -OH). The departure of the leaving group is accompanied by the [1,2]-shift of the alkyl group, which is anti to the leaving group to afford the amide after tautomerization.
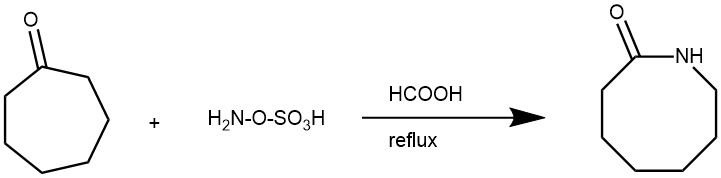
EXAMPLE 1: A 100-mL, three-necked flask is equipped with a magnetic stirring bar, a pressure-equalizing dropping funnel, and a reflux condenser connected to a nitrogen flow line. The system is dried with a heat gun while it is flushed with dry nitrogen. The reaction vessel is then cooled in a water bath while a light positive pressure of nitrogen is maintained. The flask is charged with hydroxylamine-O-sulfonic acid (8.48 g, 0.075 mol) and 95–97% formic acid (45 mL). A solution of cycloheptanone (5.61 g, 0.05 mol) in 15 mL of 95–97% formic acid is added with stirring over a 3-min period. After the addition is complete, the reaction mixture is heated under reflux for 5 hr and then cooled to room temperature. The reaction mixture is quenched with 75 mL of ice–water. The aqueous solution is slowly neutralized to pH 7 with 6 N sodium hydroxide and extracted with three 100-mL portions of chloroform. The combined organic layers are dried with anhydrous magnesium sulfate. After removal of the solvent on a rotary evaporator, the product hexahydroazocinone is purified by distillation to give 4.6 g (72%), bp 94–96°C/0.2 mm, (short-path apparatus), lit 3 bp 133–135°C/4 mm. [REF: Organic Syntheses, Coll. Vol. 7, p.254 (1990); Vol. 63, p.188 (1985)]
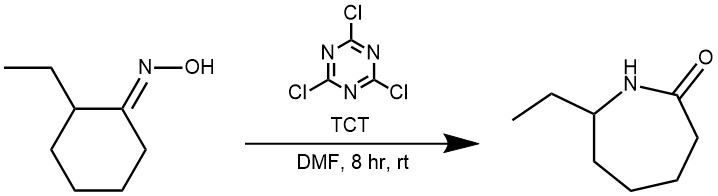
EXAMPLE 2: 2,4,6-Trichloro-[1,3,5]triazine (1.83 g, 10.0 mmol) was added to DMF (2 mL), maintained at 25 °C. After the formation of a white solid, the reaction was monitored (TLC) until the complete disappearance of TCT. Then 2-ethylcyclohexanone oxime (1.41 g,10.0 mmol) in DMF (15 mL) was added. After the addition, the mixture was stirred at room temperature, monitored (TLC) until completion (8 h). Water (20 mL) was added then the organic phase washed with 15 mL of a saturated solution of Na2CO3, followed by 1 N HCl and brine. The organic layer was dried (Na2SO4) and the solvent evaporated to yield 7-ethylazepan-2-one that was isolated without other purifications (1.41 g, 100%).[REF: J. Org. Chem. 2002, 67, 6272-6274]
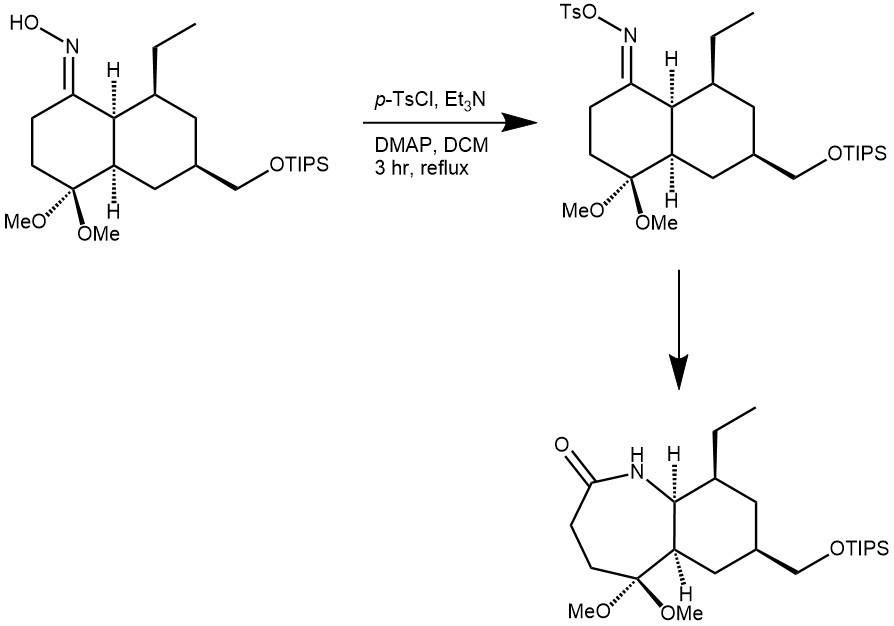
EXAMPLE 3: To a mixture of p-toluenesulfonyl chloride (203 mg, 1.07 mmol), triethylamine (0.15 mL, 1.07 mmol), and a catalytic amount of DMAP in DCM (3 mL) was added a solution of the oxime (186 mg, 0.43 mmol), and the mixture was diluted with DCM (10 mL), and the solution was washed with water and saturated aqueous NaCl, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. Chromatography of the residue on the silica gel (EtOAc-hexane, 1:3 to MeOH-CH2Cl2, 1:15) afforded 138 mg (74%) of the product as a colorless oil. [REF: Org. Lett. 2000, 2, 15, 2373–2376]
EXAMPLE 4: The oxime (1.0 mmol, 1.0 equivalent), triethyl amine (278 μL, 2.0 mmol, 2.0 equivalents) and acetonitrile (5.0 mL, 0.2 M) were sequentially added into an oven-dried reaction tube (30 mL) equipped with a stirring bar, the reaction tube was covered with a plastic stopper before the SO2F2 gas was introduced into the stirring reaction mixture by slow bubbling through a so2f2 balloon at the room temperature for 30 minutes. Then, 5% HCl (1.0 mL), 1.35 mmol, 1.35 equivalent) was added to the mixture and stirred at the room temperature for an additional 15 minutes. After that, the reaction diluted with water and extracted with dichloromethane (3*20 mL). Then the combined organic layers were washed with brine, dried over anhydrous sodium sulfate, and concentrated to dryness. The residue was purified through silica gel chromatography using a mixture of ethyl acetate and petroleum ether as eluent to afford the desired amide. [REF: European Journal of Organic chemistry, 2019, 30, 4911-4915]
REFERENCES:
- Strategic applications of named reactions in organic synthesis by Laszlo Kurti and Barbara Czako

